
| Aims and Strategy | ||||||||
| The availability of whole-genome sequences has led to the development of strategies to determine the function of previously uncharacterized genes. One such approach, whole-genome expression profiling, has been successful in identifying over 1500 genes as transcriptionally regulated during yeast sporulation (Primig et al., 2000). However, it is clear that a large portion of these genes, even those with the strongest induction, is not required for proper sporulation (Rabitsch et al., 2001). Therefore, complementary approaches are needed to assess the functional relevance of all genes in a thorough manner. The classic genetic way to assess gene function is through generation and analysis of loss-of-function mutations. In yeast, targeted disruptions of each gene in the organism were constructed in a systematic, high-throughput manner. To aid phenotypic analysis of the deletion collection, a bar-coding strategy was developed for massively parallel analysis (Shoemaker et al., 1996). We screened the collection of ~4700 homozygous diploid deletion mutants for their effects on sporulation efficiency with two goals in mind. In a comprehensive saturation genetic analysis, our first aim was to identify all functionally relevant sporulation genes in the yeast genome, both positive and negative regulators. Our second aim was to examine the correlation of expression and deletion phenotype on a whole genome scale. During the course of the study it became evident that many meiotic genes, particularly those involved in recombination and chromosome segregation, are dispensable for sporulation in our strain background. However, progeny from these meiotic mutants exhibit widespread aneuploidy and hence are largely inviable. To identify these meiotic genes, we developed a screening strategy that simultaneously identifies strains that are defective in post germination growth. The identification of mutants defective in spore germination represents the first available screen in this historically difficult field of study. | ||||||||
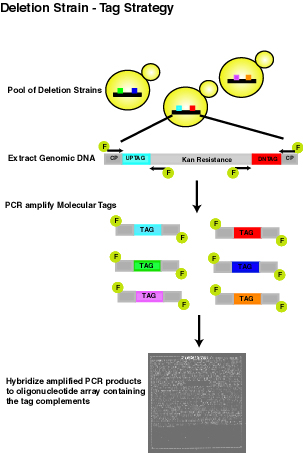 |
Individual deletion strains are quantitatively monitored in a large pool with the aid of two unique 20 base pair sequences (uptag and downtag) that serve as molecular identifiers. The relative abundance of each strain in the pool is measured on an oligonucleotide array containing the tag complements. | |||||||
 |
||||||||
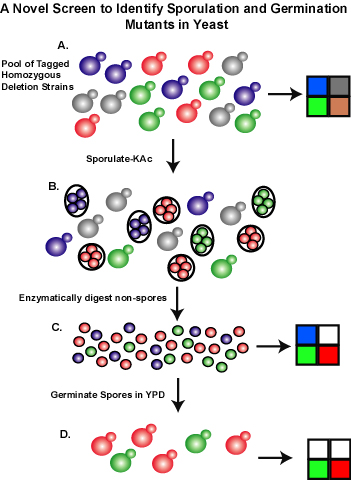
| The screen for sporulation and germination mutants is illustrated here in a simplified pool of 4 strains: grey (no sporulation), red (enhanced sporulation), blue (germination defect), and green (phenotypically normal). Prior to sporulation, an aliquot of the pool is collected and the abundance of each strain measured on an oligonucleotide array (Part A). Strain S288c sporulates to 15% efficiency, therefore non-sporulated vegetative cells were selectively degraded with zymolyase (Part B). One-half of the purified spore suspension was used to measure the presence of each strain in the population (Part C). By comparing the presporulation signal level to that post-sporulation for each strain in the pool, we could identify strains with sporulation deficiency (grey) and proficiency (red). The second half of the purified spore sample was germinated in rich media (Part D). Timepoints (3 or 4) were taken up to 20 population doublings and the presence of each strain was monitored on the tag array. A relative growth rate was calculated for each strain using a linear fit model. By comparing the post-germination growth rates of each strain to those obtained as homozygous diploids in YPD alone, we were able to identify deletion strains with a germination-specific growth defect (blue). | ||||||||
![]()
![]()
![]()
![]()
Enhanced Sporulation Efficiency Class
![]()
Post-Germination Growth Defective Class
![]()
![]()
![]()
![]()